

酵母雙雜交實驗
簡介
酵母雙雜交系統(yeast two-hybrid)以酵母遺傳分析為基礎,研究反式作用因子之間相互作用對真核基因轉錄調控影響的實驗系統。
其最有價值的應用是用 BD-X 篩選由 AD-Y 構成的 cDNA 文庫,以獲得新的蛋白質之間的相互作用,并分析研究新的基因。
本技術不僅可用于鑒定新的蛋白質相互作用,證實可疑的相互作用,確定相互作用的結構域,而且可直接獲得編碼相互作用的蛋白的基因。
原理
酵母雙雜交的基本原理是:許多轉錄因子都包含兩個相互獨立的功能結構域,即 DNA 結合結構域(binding domain,BD)和轉錄活化結構域(activedomain,AD)。
轉錄因子通過 BD 和 AD 分別與 DNA 上的特異序列結合,從而啟動相應基因的轉錄。
酵母雙雜交系統首先構建兩種反式作用因子,將蛋白質 X 與報告基因轉錄因子特異的 BD(如 Gal4-BD,LexA-BD)融合,成為釣餌(bait);蛋白質 Y 與特異的 AD(Gal4-AD,B42-AD)融合為獵物(prey)。
當編碼兩種結構域的基因在酵母細胞核內同時表達時,若蛋白 X 與 Y 之間存在非共價作用,就會使 AD 與 BD 兩結構域的上游活化序列(upstream ac-tivation sequence,UAS)相互接近,進而激活轉錄過程,使報告基因(如 HIS3、LEU 和 lacZ 等)得到表達(如圖所示)。
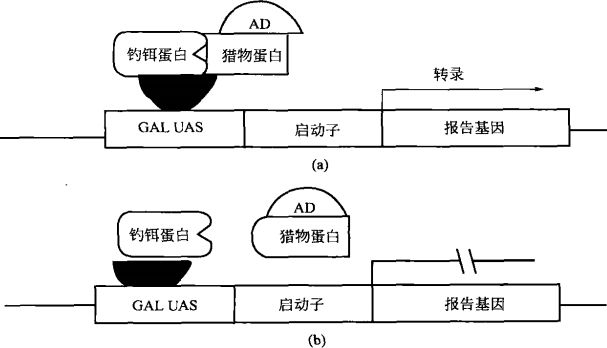
BD 與 AD 之間的連接(相互作用)能夠有效地活化轉錄。由非共價鍵連接的兩個結構域也可以通過蛋白之間的相互作用將 BD 和 AD 連接起來,從而啟動轉錄;反之,也可以通過報告基因表達來評價結構域連接蛋白之間是否存在相互作用。
用途
酵母雙雜交系統最有價值的應用是用 BD-X 篩選由 AD-Y 構成的 cDNA 文庫,以獲得新的蛋白質之間的相互作用,并分析研究新的基因。
本技術不僅可用于鑒定新的蛋白質相互作用,證實可疑的相互作用,確定相互作用的結構域,而且可直接獲得編碼相互作用的蛋白的基因。
材料與儀器
器材:
①電熱恒溫培養箱
②超凈工作臺
③臺式高速離心機、高速冷凍離心機
④恒溫水浴鍋、恒溫水浴搖床
⑤高壓滅菌鍋、除菌用濾器
⑥pH 計
⑦電轉化儀、電轉化杯
⑧旋渦混勻器
⑨多種規格錐形瓶、50 ml 離心管、15 ml 離心管、
1.5 ml Eppendorf 管、100 cm 培養皿、醋酸纖維素膜或濾紙
試劑:
①材料:酵母 AH109 或 Y187,E.coli TG1,cDNA 文庫
②胰蛋白胨、酵母抽提物、瓊脂、葡萄糖、各種氨基酸、酵母氮堿
③ NaCl、PEG4000
④吡喃半乳糖苷衍生物(5-bromo-4-chloro-3-indolyl-α-D-galactopyranoside,X-a-gal)或 X-β- gal
⑤ 1 mol/L 3-氨基三唑(3-amino-1,2,4-triazole,3-AT)
⑥魚精 DNA
⑦ DMSO、甘油
⑧醋酸鋰
⑨ SDS、Lyticase
⑩酚-氯仿(1:1)、無水乙醇等
步驟
酵母雙雜交的基本過程可分為如下幾步:
1.酵母感受態細胞的制備和載體的轉化(小規模轉化)
(1)從 YPD 平板上挑取生長 1~2 周、直徑 2~3 mm大小的新鮮酵母 AH109 單克隆,接種到 1 ml 的滅菌去離子水中,劇烈振蕩以分散酵母團。
(2)轉移上述酵母細胞到 30 ml YPD 培養液中。
(3)30 ℃、250 r/min 振搖培養過夜,直到穩定期(OD600>1.5)。
(4)取適量上述培養物轉入150 ml YPD 培養液中至 OD6oo 達到 0.2~0.3。30 ℃、250 r/min振搖培養至 OD600 達到 0.5±0.1。
(5)轉入 50 ml 離心管中,室溫 4000 g 離心 5 min,棄上清液。在25~50 ml 滅菌TE或蒸餾水中劇烈振蕩重懸細胞。重復離心棄上清。
(6)在 0.75 ml 1xTE/LiAc 中重懸沉淀,此即為酵母感受態細胞,冰浴備用。
(7)在 1.5 ml Eppendorf 管中加入下列成分并振蕩混勻。
(8)加入酵母感受態細胞 0.1 ml,振蕩混勻。
(9)加入 0.6 ml 滅菌的 PEG/LiAc 溶液并高速振蕩混勻。
(10)30 ℃、200 r/min 振搖培養 30 min。
(11)加入 70 μlDMSO,上下輕輕顛倒混勻(不能劇烈振蕩)。42 ℃熱休克 15 min,立即冰浴冷卻 2 min。
(12)4000 g 離心 5 min,盡可能棄盡上清液。在0.5 ml 的 1xTE(pH7.5)中重懸細胞備用。
2.轉化混合物的鋪盤和篩選
(1)按圖 12-5 所示的方法將上述轉化混合物涂布于適當培養基平板上(直徑 100 mm的培養皿)。一個新的釣餌與文庫的結合,很難去預測最適的方法。
因而,各 鋪1/3 的轉化物于不同的選擇培養基上:低嚴謹度(SD/-Leu/-Trp),中嚴謹度(SD/-His/-Leu/-Trp)和高嚴謹度(SD/-Ade/-His/-Leu/-Trp/X-a-gal)的培養基上。
(2)30 ℃ 倒置培養至長出克隆,其中在 X-a-gal 培養基上顯藍色的克隆為陽性。也可不加 X-a-gal,按步驟(3)進行 β 半乳糖苷酶濾膜印跡法篩選。
(3)β 半乳糖苷酶濾膜印跡實驗確定陽性克隆
①準備 Z 緩沖液/X-β-gal 溶液。
②用鑷子將一干燥的滅菌硝酸纖維濾膜(或濾紙)置于上述菌落盤上,輕輕用鑷子壓膜以便克隆黏附到濾膜上。
③當濾膜均勻地潤濕后,小心地將濾膜從培養基上取出,放入液氮中。
④在完全浸入液氮中約 15 s 后,將濾膜取出并將有克隆的一面朝上放入潔凈培養皿(或皿蓋)中。
⑤加 1~2 mlZ 緩沖液/X-β-gal 溶液,至濾膜全部潤濕。
⑥30 ℃ 靜置孵育。在8h內檢查藍色克隆的出現。
3.陽性克隆的鑒定和證實
(1)將半乳糖苷酶陽性的克隆劃線接種于 SD/-Leu/-Trp/X-α-Gal 盤上;或者接種于 SD/-Leu/-Trp 盤上,長出克隆后再進行β半乳糖苷酶濾膜印跡法鑒定。同時儲存陽性克隆于 -70 ℃。
(2)30 ℃ 倒置培養 4~6 d。觀察克隆顏色的變化或用 β 半乳糖苷酶濾膜印跡測定來顯示顏色的變化。
(3)重復上述步驟(1)~(2)2~3次。
4.酵母質粒的分離
(1)在陽性克隆酵母盤上擴增一定數量的酵母,刮下約 30 μl,加 50 μlTE(pH7.0),旋渦振蕩以懸浮細胞。
(2)加 10 μl Lyticase 裂解液(5 U/μl),旋渦振蕩或吸管反復吹打混勻。
(3)37 ℃、250 r/min振搖孵育 1 h。
(4)加 10 μl 20% SDS,旋渦振蕩 1 min。
(5)冷凍(-20 ℃)/融化一次,旋渦振蕩以充分裂解細胞。
(6)用 TE 補足 200 μl。
(7)加 200 μl 酚-氯仿(1:1),高速旋渦振蕩 5 min。
(8)10000 g 離心 10 min,轉移水相于新的 Eppendorf 管。
(9)加 1/10 體積的 5 mol/L NH4Ac 溶液和兩倍體積的無水乙醇。
(10)-70 ℃ 冷凍 1 h。
(11)10000 g 離心 10 min,棄上清液。
(12)干燥 DNA 沉淀,溶解于 20 μl 滅菌水中。
5.酵母質粒的電轉化
(1)將2~5 μl 酵母質粒加入 40 μl 感受態酵母細胞中,混勻,冰浴。
(2)混合液轉入冰預冷的電轉化杯中,設置適當的參數(如 1.8 kV,25 μF,200 Ω),電擊。
(3)迅速將細胞液轉入 1 ml SOC 培養液中,輕輕混勻。37 ℃、250 r/min 振搖孵育 1h。
(4)4000 g 離心 5 min,棄上清液,保留 100 μl 左右培養液,輕輕重懸后涂布于 LB/Amp 培養皿平板,37 ℃ 倒置培養過夜。
細菌質粒按常規方法抽提。將抽取的質粒進行小規模轉化實驗以證實陽性克隆與已知基因的相互作用。
6.陽性克隆的分類
用插入 cDNA 片段兩端的限制性核酸內切酶酶切質粒,瓊脂糖凝膠電泳鑒定插入片段的大小,并以此進行分類。
7.代表性克隆與釣餌的共轉化
取每類中的代表性克隆與釣餌共轉化以證實在酵母中是否確實相互作用(方法同小規模轉化實驗)。
8.對證實的陽性克隆質粒進行測序。
9.測得的序列進行 Blast 比較。如為未知的 cDNA 序列,可進一步進行其他的生物信息學分析(www.ncbi.nlm.nih.gov/blast)。
10.獲得的相互作用的蛋白質對還需用其他方法證實,如免疫共沉淀、GST-pull down 等。
注意事項
(1)小規模轉化實驗常用于:證實不能自我活化報告基因的 DNA-BD/釣餌,確證 DNA-BD/釣餌對宿主是否具有毒性,對照實驗以及順序性轉化時用于轉化 DNA-BD/釣餌。
順序轉化即 DNA-BD/釣餌質粒先通過小規模轉化進入酵母,然后再將AD 融合的庫質粒轉人選擇的酵母克隆中。
(2)一般抽提的酵母質粒由于 DNA 含量少而且混有雜質,用化學轉化方法很難獲得轉化的細菌克隆,故適合用電轉化法。
電轉化時,應嘗試不同的設置以找到合適的參數。
(3)在 β 半乳糖苷酶濾膜印跡實驗中,克隆應有 1~3 mm 直徑大小。如果每盤上克隆只有幾個,可將克隆集中到一主盤上來測定。
為了方便確定陽性克隆,可用尖頭鑷子在濾膜和培養基上扎幾個小洞(不對稱分布)做位置標記。
